Accelerate the process of drug discovery with the use of a new neural network
How a new neural network can predict and understand molecules in a more efficient way
13 October 2021
The network operates on molecular data or any type of n-dimensional point cloud representation. The research presents a different technique where equivariance can be achieved in a more simple, more efficient way which works better. Equivariance in the context of this research means that applying a transformation on a molecule does not have any effect on the translating and rotating it. Eventually, predictions about new molecules can be made and a better understanding about already existing molecules can be achieved.
Understanding molecules
Molecules are everywhere around us, and these molecules are composed by atoms interacting with each other and sticking together. As the interactions between these atoms are scaled up, they may result in more complex structures such as proteins, cells or life organisms. The water you drink and the device you are reading this from is made out of molecules as well. Victor and Emiel, both PhD students from the Amsterdam Machine Learning Lab (AMLab), and co-author Professor Max Welling presented a new neural network which takes a look at molecule data in a 3-dimensional representation environment. This is done by the use of deep learning, where a lot of data is used to train the computer to think like a human would do. Eventually the trained neural network is able to make predictions about molecular properties, such as if the molecule could have toxicity, homo-lumo energy gap or binding affinity which is important for further work with the molecule. It can also compute a transformation on any arbitrary n-dimensional point cloud representation (e.g. molecules). By doing so, molecule generation can accelerate the process of drug discovery and material discovery. For example, in 2020 a group from Massachusetts Institute of Technology (MIT) generated a new antibiotic which they called Halicin using neural networks. The drug made the killing of the most problematic disease-causing bacteria possible.
The network
To operate on the molecular data, the network functions as a message passing algorithm that propagates information among atoms. After a certain number of propagation steps, the network tries to make a prediction about the molecular properties, or it can compute a transformation on the molecule. Predicting molecular properties is of interest in material or drug discovery because different properties may lead to different chemical reactions. As a consequence, new molecules can potentially be generated, filtering on some desired or undesired properties. This all could be important during the drug discovery process.
Molecular invariance
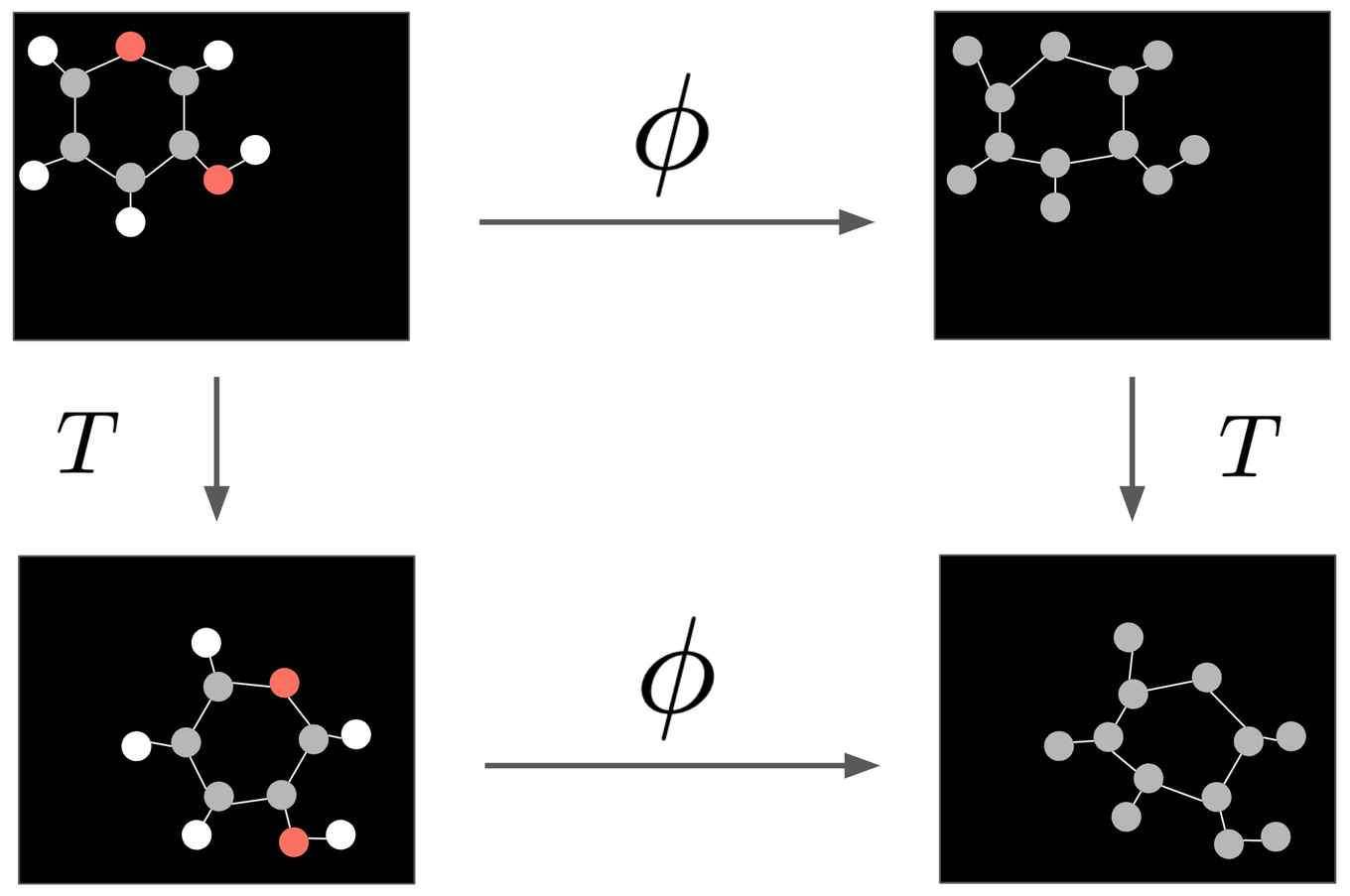
To make the right prediction about molecular properties, it is desired to satisfy some constraints to the neural network. For this network invariance to translations and rotations is desired. The position and orientation of a molecule should not modify its intrinsic properties. When performing a transformation on the molecule, equivariance is desired, this means that translating or rotating the molecule before applying the transformation is equivalent to doing so after applying the transformation. The presented network satisfies these constraints, which means that when given a certain position and orientation, the model automatically generalizes to all its possible translations and rotations. A visual example of this phenomena is presented in the figure below. The used technique satisfies the equivariant constraints in a simpler and more efficient way than previous methods by only considering the relative differences and distances between atoms as positional information.
Evaluating the model
The method is evaluated in modelling dynamical systems, representation learning in graph autoencoding and predicting molecular properties in the QM9 dataset. The method reports best or competitive performance in all experiments while remaining more efficient in running time than previous methods.
Link to the original paper
Victor and Emiel will continue with their research in collaboration with researchers from the University of Oxford. The Robert Bosch GmbH is acknowledged for financial support.


