Rational optimization of catalysts for asymmetric hydrogenation
Tuning hydrogen bonding in supramolecular rhodium complex improves reaction rate and selectivity
25 september 2017
The generation of chiral compounds is key to many chemistry related fields, including pharma, fragrance, materials and fine chemicals. The preparation of enantiopure compounds therefore is of crucial importance. These can be obtained through entioselective synthesis, employing asymmetric catalysis that favours the formation of the desired chiral compound over its counterpart.
Developing new catalysts for new asymmetric conversions in general requires a lot of trail-and-error, as computational prediction of the selectivity of the catalysts is too challenging. Researchers at HIMS now report a strategy that brings chemistry one step closer to a more efficient catalyst development by rational design.
Computational study
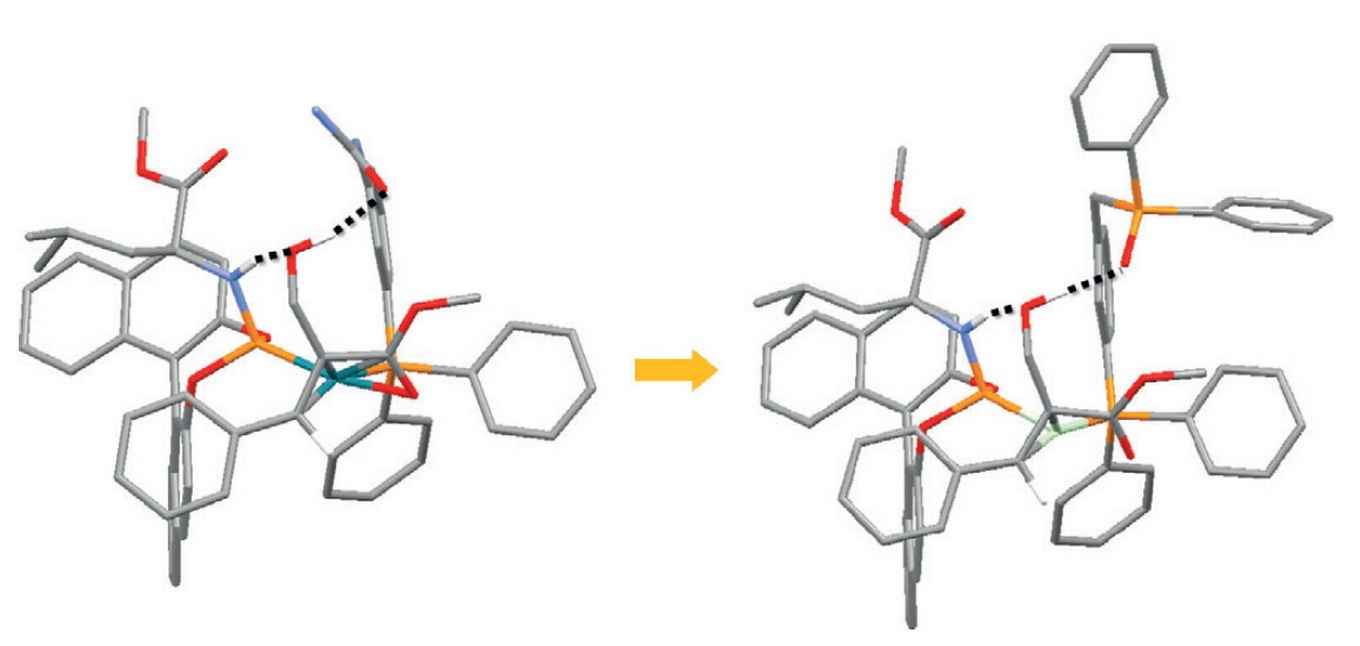
At the homogeneous and supramolecular catalysis research group of professor Reek supramolecular catalysts have been developed where a rhodium complex acts as a selective catalyst for asymmetric hydrogenation. From a detailed study the Amsterdam researchers were able to establish the crucial role of hydrogen bonding between the catalyst and the substrate. The experimental data were used as input for a computational study to exactly determine the reaction pathway and the specific role of the hydrogen bonds. As it turns out, the hydrogen bonds not only lead to better pre-association of the substrate, but also lower the transition state of the rate limiting step.
Based on this information a new catalyst was developed by replacing a urea group for a phosphine oxide, thus optimizing the hydrogen bonding. According to DFT calculations this should result in a faster reaction, because of a lower energy pathway, which was indeed confirmed by experiments. In addition, the catalyst is also more selective and more robust at elevated temperatures.
With this study the researchers show that supramolecular interactions between substrate and catalyst are of crucial importance, and, importantly, catalyst can be optimized in silico by just 'zooming in' on these interactions. Thanks to the Amsterdam research, chemists now have a new tool in hand to efficiently design new catalysts for new asymmetric conversions.

Article
Daubignard, J., Detz, R. J., Jans, A. C. H., de Bruin, B. and Reek, J. N. H. (2017), Rational Optimization of Supramolecular Catalysts for the Rhodium-Catalyzed Asymmetric Hydrogenation Reaction. Angew. Chem. Int. Ed.. DOI:10.1002/anie.201707670